
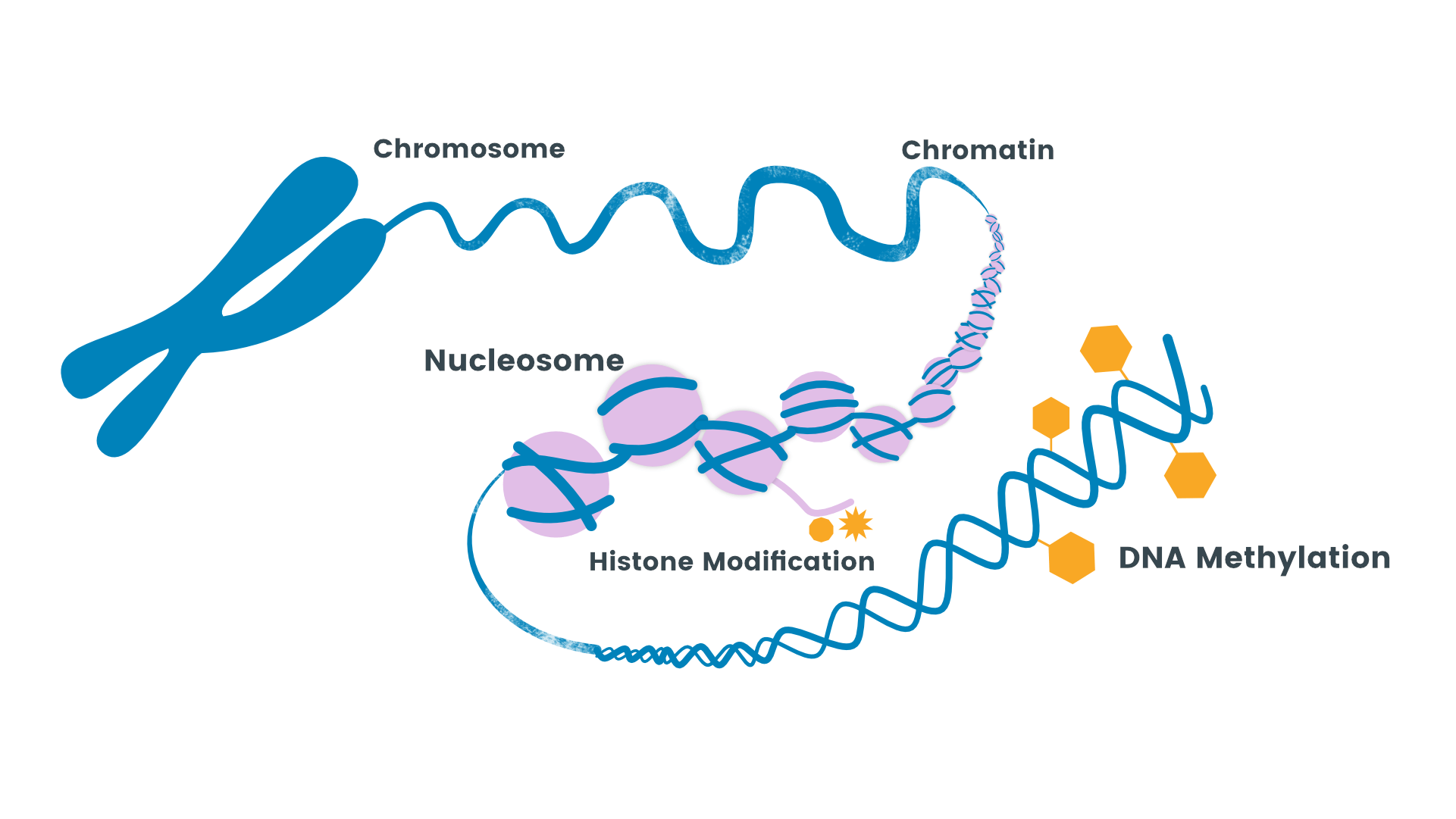
La metilación del ADN juega un papel central en la regulación y expresión génica. La metilación es un mecanismo epigenético que bloquea la expresión de genes. Es por ello que constituye un proceso clave en la impronta genética, el desarrollo embrionario o la inactivación del cromosoma X. Por todo ello, muchos científicos han asociado un estado anormal del estado de metilación de un gen a diferentes patologías.
- Regulación de la metilación del ADN mediante ADN metiltransferasas
- Implicaciones de la metilación del ADN
- Metilación del ADN y enfermedad
- Análisis de metilación mediante EpiTYPER de Agena Bioscience
Regulación de la metilación del ADN mediante ADN metiltransferasas
La metilación del ADN implica la adición de un grupo metilo al 5º átomo de carbono del anillo de citosina, lo que resulta en 5-metilcitosina o 5-mC. Estos grupos metilo inhiben la transcripción al ocupar el surco principal del ADN. 5-mC constituye aproximadamente el 1,5% del ADN genómico. La metilación del ADN es catalizada por ADN metiltransferasas (DNMT). Existen tres tipos de DNMT: DNMT1, DNMT3a y DNMT3b para establecer y mantener los patrones de metilación del ADN.
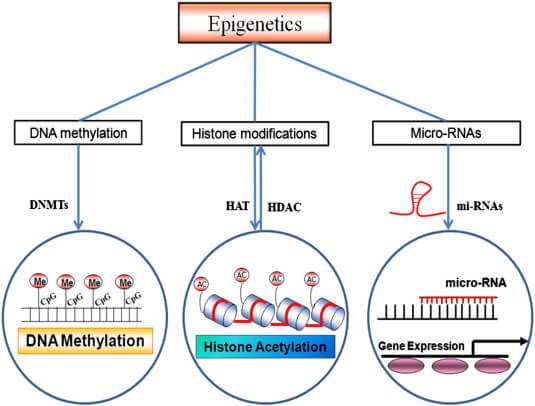
La proteína DNMT1 está involucrada en el mantenimiento de los patrones de metilación del ADN que ya están establecidos, mientras que DNMT3a y DNMT3b parecen estar involucradas en el establecimiento de nuevos patrones de metilación del ADN. Es decir, en ciertos tipos de cáncer, esto puede ser diferente, ya que tanto DNMT1 como DNMT3b pueden participar en el mantenimiento de la hipermetilación en las células cancerosas.
La metilación del ADN es una modificación epigenética reversible. La eliminación de un grupo metilo o la desmetilación es necesaria para reprogramar los genes y, por lo tanto, también es importante en el crecimiento de tumores. La desmetilación es catalizada por enzimas llamadas desmetilasas del ADN.
Implicaciones de la metilación del ADN
El papel de la metilación del ADN en la expresión génica varía según en que reino de organismos nos encontremos. Por ejemplo, la metilación de CpG se distribuye de manera bastante global en los mamíferos, mientras que entre los invertebrados, el patrón de metilación generalmente ocurre en forma de «mosaico», con regiones de ADN fuertemente metiladas intercaladas con regiones que no están metiladas. En plantas, el 50% de los residuos de citosina están en un estado metilado, mientras que en los hongos, solo se metilan secuencias repetidas de ADN.
La importancia de la metilación del 5-mC, es una modificación epigenética clave y ámpliamente reconocida. De tal forma que en algunos procesos, es probable que una disminución en la metilación global del ADN (hipometilación del ADN) sea el resultado de la deficiencia de grupos metilo causada por diversos factores ambientales y se ha sugerido como un marcador molecular en muchos procesos biológicos, incluido el cáncer.
Metilación del ADN y enfermedad
Dado que la metilación del ADN desempeña un papel tan importante en la expresión génica, parece obvio pensar que una metilación defectuosa podría tener consecuencias devastadoras, incluida la aparición de enfermedades. Existen muchos estudios que han examinado la asociación entre los errores de metilación y enfermedades como el cáncer, la distrofia muscular, el lupus y varios defectos de nacimiento. Se espera que esto proporcione información valiosa para mejorar la comprensión, el tratamiento y la prevención de estos trastornos.
Muchos estudios se han focalizado en la conexión entre la metilación del ADN y los genes supresores de tumores y el cáncer. Se ha demostrado que la hipermetilación del ADN silencia los genes supresores de tumores en las células cancerosas.
Por el contrario, los genomas de las células cancerígenas han mostrado presentar un estado de hipometilación en general. Sin embargo, los genes involucrados en la invasión de las células tumorales, reparación del ADN y los genes implicados en la regulación del ciclo celular suelen estar hipermetilados. Todos estos procesos conducen a un silenciamiento de la expresión y comportan metástasis.
La detección de la hipermetilación en algunos cánceres, como el cáncer de colon, es posible en una etapa temprana del curso de la enfermedad y puede servir como un biomarcador valioso para la enfermedad.
Análisis de metilación mediante EpiTYPER de Agena Bioscience
La tecnología de análisis de metilación del ADN, EpiTYPER MassARRAY, proporcionada por Agena Bioscience (anteriormente Sequenom Inc.), es uno de los mejores métodos cuantitativos disponibles en el mercado para el análisis de la metilación del ADN. EpiTYPER es un método de secuenciación de bisulfato basado en espectrometría de masas (MALDI-TOF) que permite el análisis de metilación del ADN específico de región de forma cuantitativa y de alto rendimiento. La tecnología es capaz de interrogar entre decenas y cientos de muestras y regiones CpG en amplicones de entre 200 a 600 pb y detecta hasta un 5% de diferencias en el estado de metilación.
Flujo de trabajo: del diseño del ensayo a los resultados
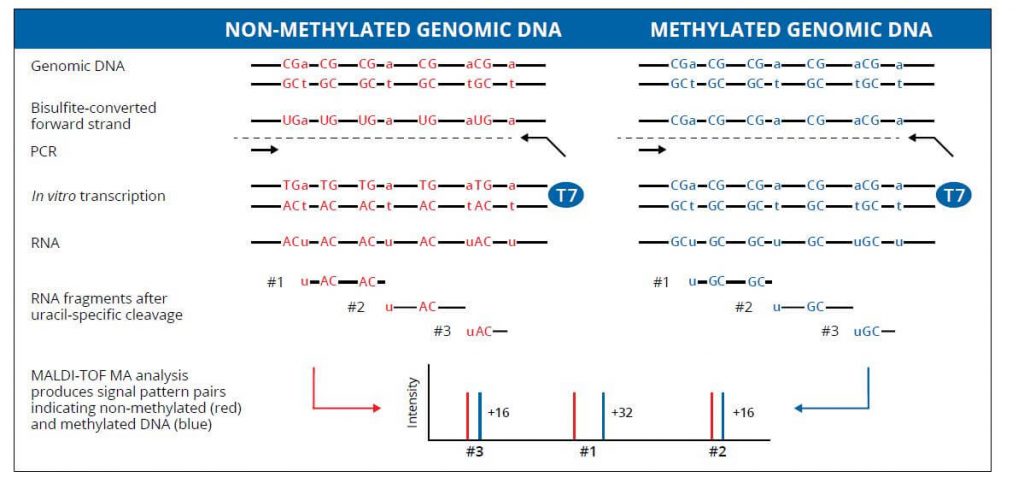
La bioquímica EpiTYPER comienza con el tratamiento con bisulfito del ADN genómico, seguido de la amplificación por PCR de las regiones de interés. El cebador inversos (reverse primer) contienen una secuencia promotora para T7 polimerasa. A continuación, se realiza la transcripción de ARN in vitro, seguida de la escisión del ARN específico de la base. Finalmente, los productos de escisión se analizan utilizando espectrometría de masas MALDI-TOF (analizador MassARRAY). Los fragmentos de citosina metilados y no metilados en el ADN genómico original se distinguen utilizando el software EpiTYPER.
Paso 1: Diseño del experimento
EpiDesigner es una herramienta online para el diseño automatizado de experimentos de metilación de ADN utilizando el sistema MassARRAY. De forma resumida, debemos ingresar las secuencias a estudiar y el software determinará el diseño de los primers para la cobertura del ADN más completa. Además de las secuencias de cebadores optimizadas, EpiDesigner brinda una interpretación gráfica fácil de leer de los amplicones diseñados sobre sus regiones objetivo, así como anotando distintos sitios CpG cubiertos por los ensayos.
Paso 2: Tratamiento Bisulfito
El ADN genómico es tratado con bisulfito, lo que conduce a una conversión de todas las citosinas no metiladas, mientras que las citosinas metiladas no se ven afectadas.
Paso 3: PCR, Transcripción in vitro y escisión del ARN
El ensayo EpiTYPER se inicia con una PCR que utiliza un cebador inverso (reverse primer) que contiene la secuencia del promotor polimerasa T7 para amplificar la región diana y preservar los cambios de secuencia inducidos por bisulfito.
Después del tratamiento con fosfatasa alcalina (SAP), para descartar los nucleótidos no incorporados, se realiza la transcripción in vitro, el promotor T7 agregado durante la PCR se usa para transcribir el producto de la PCR de la cadena inversa, produciendo un producto de ARN monocatenario. Este producto de ARN se escinde específicamente con RNasa A en los residuos de uracilo generados.
Paso 4: Adquisición de datos e análisis
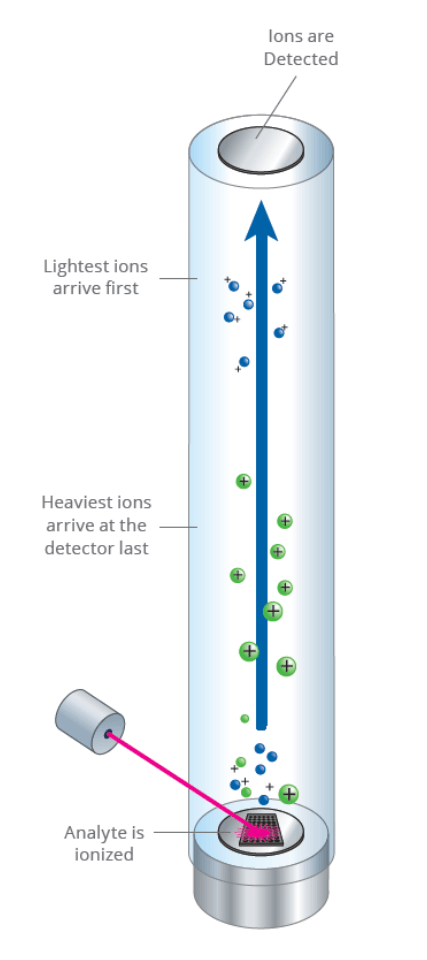
EpiTYPER de Agena Bioscience dispensa los analitos en una matriz llamada SpectroCHIP®. Luego, el chip se coloca en el espectrómetro de masas MALDI-TOF para la adquisición de datos, que generalmente requiere de 15 a 60 minutos. Los resultados se cargan automáticamente en una base de datos para el análisis de datos con el software.
Los amplicones generados se separan gracias al tiempo que tardan en llegar al detector, es decir, determinan el tiempo de vuelo de las moléculas. El tiempo de vuelo aumenta con mayor masa. Un fragmento que contiene uno o más dinucleótidos CpG se denomina unidad CpG. Si se metiló un dinucleótido CpG y se protegió de la conversión con bisulfito, el fragmento de ARN correspondiente, la unidad CpG, tendrá una masa de 16Da más pesada cuando se metiló el dinucleótido CpG, dando como resultado un cambio de 16Da en el espectro de masas. La señal detectada por el espectrómetro de masas para cualquiera de los fragmentos es proporcional al número de fragmentos. El número de fragmentos se cuantifica por el área de superficie de los picos correspondientes en el espectro de masas. El porcentaje de metilación del ADN de un CpG dado se calcula dividiendo el área de superficie del pico que representa el fragmento metilado por el área de superficie total de los picos del fragmento tanto metilado como no metilado.
Principales características y Ventajas
| Eficiente | Preciso | Sensible | Rentable | Flujo de trabajo sencillo |
|
|
|
|
|


Deja tu respuesta