
RNA expression profiling of biological models to identify which genes are expressed and which pathways are active in biological systems under different conditions is critical to unraveling how genes control biology. For years, qRT-PCR has provided a robust and reliable approach to assessing transcript levels of any gene. However, the qRT-PCR assay is limited to a few objectives. Approaches using oligonucleotide probe arrays have been developed to address this throughput limitation, but microarray RNA expression analysis does not provide robust measurements of expression levels that qRT-PCR provides. In addition, indirect fluorescence detection of transcripts hybridized to arrayed oligonucleotides has the problem of background noise and signals caused by cross-hybridization.
With the advent of affordable and accessible next generation sequencing (NGS) technology, complete sequencing of cDNA made from RNA isolated from biological models of interest became much more feasible. RNA-seq approaches have overtaken microarrays as the method of choice for genome-wide expression analysis.
Although it provides an accurate readout of the transcripts present in biological samples, there are also limitations of RNA-seq. It can be a difficult test to perform for a large number of samples. In addition to RNA extraction, sample preparation, especially for groups primarily interested in protein expression genes, requires mRNA enrichment. For blood samples, additional work is required to deplete the levels of the beta-globin gene. Even after this preparative work on the RNA is completed the assay involves cDNA synthesis of the total transcriptome, followed by generation of NGS libraries from the total complement of fragmented cDNA. NGS analysis of these complex samples requires significant sequencing depth (usually 25 to 50 million reads), as well as involved deconvolution and bioinformatic processing to estimate the copy number of each gene from large numbers of variable gene fragment reads. Driver map offers an alternative to RNA-seq.
Overcoming the Limitations of RNA-seq – Genome-Wide Targeted RNA Sequencing for Expression Profiling
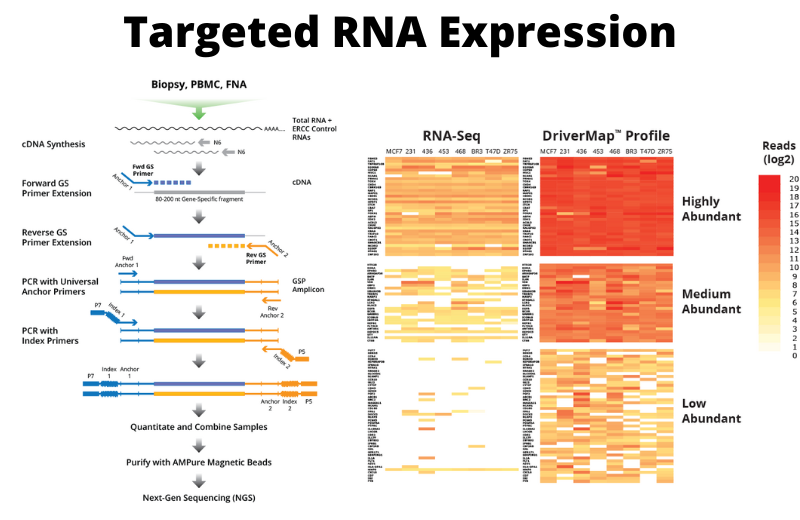
An alternative to whole transcriptome sequencing is to use a targeted RNA sequencing approach. This is the basis of the DriverMap targeted RNA expression assay developed by Cellecta. Rather than reverse transcribing the entire transcriptome, the DriverMap approach combines multiplex RT-PCR amplification to amplify a defined and conserved 80- to 250-base segment of the transcript from each target gene, and then uses NGS sequencing to quantitatively assess the abundance levels of each of these transcript amplicons.
There are significant advantages in targeting, amplifying, and sequencing carefully selected discrete loci of interest in the expressed transcriptome. From a procedural perspective, the use of gene-specific primers that target each gene of interest for reverse transcriptase and PCR reactions eliminates the need to prepare RNA to remove unwanted sequences such as ribosomal RNA, beta-globins, or other non-coding RNA.

Only transcript sequences that match the targets of interest are amplified. As a result, only total RNA is needed for the assay, and since it is based on PCR, the requirement is low. As little as 10 pg of total RNA from a single cell lysate is sufficient to detect most transcripts.
DriverMap RT-PCR’s targeted approach also greatly simplifies the cDNA library to be sequenced. A multiplex assay targeting all 19,000 human protein-coding genes yields, at most, only 19,000 amplicons for sequencing. The NGS read depth required to reliably read several thousand specific amplicons is much less than the depth required for the entire transcriptome. As a result, the targeted approach detects transcripts with lower level of expression more consistently and reliably than other RNA-seq approaches and with much less sequencing.

In addition to requiring fewer reads in NGS, analysis of the results is much simpler using DriverMap, as each cDNA target region provides a reference sequence with which to directly compare sequencing reads. There is no need to estimate gene copy number from assembles cDNA fragments. With targeted RT-PCR, the readout levels of each amplicon directly correlate with the expression levels of the target transcript.
Development of primers for targeted RNA analysis: DriverMap
The key to the targeted RNA approach is the efficiency and specificity of the multiplex PCR target amplification step. Numerous factors that can be tolerated in simple PCR reactions, such as nonspecific binding, primer binding inefficiency, and primer-primer interactions, can cause significant problems in a multiplex PCR environment, which amplifies many thousands of sequences at a time at the same reaction.
Development of the DriverMap assay required sophisticated primer design work and an iterative empirical testing process. For this, a bioinformatic primer design pipeline was developed and the experimental validation of thousands of PCR primers was carried out in a highly complex multiplex reaction. This approach enabled the generation of an optimized genome-wide multiplex PCR assay for all 19,000 human protein-coding genes. Initially, primers were selected and screened for various characteristics. For example, all primers are selected to have GCA-rich sequences (with minimal T content) to reduce primer dimer formation in the multiplex PCR assay.

Additionally, primers are selected for high specificity, high Tm, and small amplicon size with a balanced nucleotide distribution to ensure robust NGS.

In addition to bioinformatic screening, each set of primers also had to be tested experimentally. For each target mRNA, the best 5–20 primers were synthesized and run in several multiplex RT-PCR reactions using a pool of universal control human tissue/cell line RNAs, mouse negative control RNAs, and positive controls. A set of primers targeting the conservative portions of different mRNA isoforms with the highest efficiency, sensitivity, and specificity for each mRNA was selected. Furthermore, for highly abundant transcripts, primers with low specific efficiency were selected, which allowed us to solve the problem of oversequencing of highly expressed transcripts.
While DriverMap Targeted RNA Profiling offers numerous benefits over standard RNA sequencing, it is not a replacement for the broader RNA-seq application. The DriverMap assay requires knowledge of the transcripts of interest. Since cDNA synthesis and PCR primers target specific regions in expressed RNA from known genes, it is not a useful approach to identify novel transcripts nor a good approach to investigate alternative isoforms of particular genes. Furthermore, the assay, as configured, targets genes that code for proteins. It will not detect non-coding RNA or other non-targeted transcripts. However, custom assays can be developed for these purposes.
Overall, it provides a convenient and effective method of identifying which pathways are active in biological samples of interest. As a PCR assay, it is particularly effective when samples are limited with very small amounts of RNA. Furthermore, due to the ability to target selected sets of genes, it may be especially useful for quantifying cells or biomarkers expressed at very low levels. Custom assays can also be designed to specifically target low-level genes of interest. In particular, for researchers interested in measuring the transcript levels of specific cell fractions in heterogeneous samples, such as immune cells in tumor samples, this ability to detect low-expressing genes may be crucial. They offer pre-built panels for humans and mouse, as well as custom dashboards.
If you want more information about DriverMap technology, or want to know if we can make one for your needs, do not hesitate to contact us:



Leave a reply