
Are you starting Immunohistochemistry experiments for the first time? Comfortable with the method, but searching for few extra tips? We are going to have an answer to this one and some others.
- 1. Tissue preparation
- 2. Antigen retrieval
- 3. Sample handling
- 4. Permeabilization
- 5. Blocking
- 6. Choosing your primary antibody
- 7. Control Antibodies
- 8. Antibody dilutions
- 9. Washings
- 10. Detection
Immunohistochemistry, or IHC, allows you to visualize proteins in intact tissue retaining its microstructure, or as I like to say: ‘real life’. One of the advantages of this ‘real life’ visualization is that it allows for comparison between healthy and diseased tissues, thus the application is invaluable to both scientist and pathologist alike. The IHC protocol is straightforward, but contains many steps that may require initial optimization to ensure specific antibody binding and optimal visualization of the target protein. Here, we outline some tips for each of these steps and highlight which may need a little tweaking.
1. Tissue preparation
Tissue samples can be frozen or fixed. Freezing the sections generally maintains the conformation of the target antigen allowing superior antibody binding, but small ice crystals may form in the tissue and these sections may not be a good choice for long-term storage. 
Fixed and embedded tissue is a better alternative if you wish to hang on to your slides a little longer. The most common method of tissue fixation and embedding is formaldehyde fixation with paraffin embedding (FFPE). (It is possible to store FFPE embedded biopsies indefinitely at room temperature – making them an important resource for historical studies in medicine!).
Fixing tissue sections, before staining, with ice cold 50:50 methanol-acetone (MeAc) or 4% paraformaldehyde (PFA) may enhance antibody binding. MeAc, for example, dissolves cytoplasmic proteins allowing better visualization of membrane bound proteins.
2. Antigen retrieval
Formaldehyde fixing causes cross-linking of proteins within the tissue, maintaining tissue morphology but denaturing the epitopes recognized by the antibody. Therefore, antigen retrieval is usually carried out before staining FFPE sections to unmask hidden or denatured target epitopes. This can be heat-induced or enzymatic. These factors (temperature and pH) should be tested. It is a good idea to test a ‘battery’ or panel of antigen retrieval methods for each new antibody and tissue combination to determine optimal staining and rule out any non-specific background staining.
A good starting point is boiling in citrate buffer at pH 6.0 in the microwave. Antigen retrieval is not needed for frozen sections, but the sections should be air dried for at least an hour prior to staining. De-waxing of FFPE sections in xylene and alcohol must be carried out prior to antigen retrieval and staining; following de-waxing, sections are rehydrated in decreasing levels of alcohol and then water.
3. Sample handling
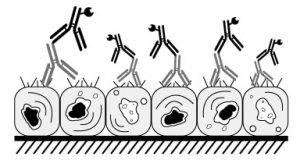
Once sections are de-waxed and rehydrated it is important not to let them dry out! So use a specialized IHC staining tray to keep them in during incubations – many vendors sell these, they help keep tissue sections in a humidified environment. Alternatively, use a tray with tissue soaked in water.
Top tip: Use a PAP pen– a special marking pen that provides a thin film-like hydrophobic barrier when drawn around a specimen – to keep solutions close to the sections once the permeabilization step has been carried out. Its use is not entirely essential but can be useful.
4. Permeabilization
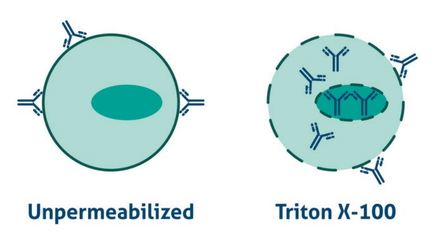 For most proteins, it is a good idea to add a permeabilization step to your protocol. Permeabilization may not be necessary for transmembrane proteins whose epitopes are in the extracellular region. Permeabilization involves incubation with a detergent (e.g. 0.1% Triton-X100 in PBS). Detergents can also act as surfactants, breaking through some of the protein cross-linking that occurs during formaldehyde fixing, therefore aiding antibody binding to the correct epitope and reducing non-specific hydrophobic interactions. Top tip: If staining isn’t working, try including detergent such as Triton at a lower level in all solutions (particularly for FFPE staining).
For most proteins, it is a good idea to add a permeabilization step to your protocol. Permeabilization may not be necessary for transmembrane proteins whose epitopes are in the extracellular region. Permeabilization involves incubation with a detergent (e.g. 0.1% Triton-X100 in PBS). Detergents can also act as surfactants, breaking through some of the protein cross-linking that occurs during formaldehyde fixing, therefore aiding antibody binding to the correct epitope and reducing non-specific hydrophobic interactions. Top tip: If staining isn’t working, try including detergent such as Triton at a lower level in all solutions (particularly for FFPE staining).
5. Blocking
The blocking solution binds to non-specific binding sites within the tissue, thus preventing non-specific binding of the primary and secondary antibodies to tissue components. Sections are incubated with an ‘innocuous protein solution’ such as bovine serum albumin (BSA) or serum from the host of the secondary antibody – I usually start with 3% BSA solution for 20-30 minutes. Proteintech also routinely uses 3% BSA.
But can also use 5-10% goat serum as an alternative (as the secondary antibody it employs is raised in goat).

6. Choosing your primary antibody
Your choice of primary antibody should depend on factors such as expression levels of the protein-of-interest and any other antibodies used in the case of co-localization. Antibodies from different species must be used in the case of the latter so that different secondary antibodies with unique conjugates can be used to distinguish between them. You must also choose whether a monoclonal or polyclonal is more suited to your needs. Monoclonal antibodies give a highly specific signal whereas polyclonal antibodies can give a brighter one, particularly if expression levels of the protein of interest are low.
7. Control Antibodies
If you wish to carry out controls to test the specificity of primary antibody binding, a few options are set out below:
- The ideal control would be to use tissue that does not contain the protein of interest, for example: tissue from a knock-out mouse or cells where the target protein is knocked down i.e. with siRNA
- Western blotting is used to check that the antibody is detecting a protein of the right size. Multiple bands on a Western blot suggest non-specific antibody binding.
- Antibody specificity can be checked by pre-incubation of the primary antibody with the antigenic peptide (an adsorption test). This step is not necessary if the antibody is affinity-purified, as the specific peptide sequence is used for the protein purification.
- If the antibody is custom made, carry out Western blotting with serum from the various steps of the production process to determine whether the band representing your protein is detected by the correct fractions.
 [/vc_column_text][/vc_column][/vc_row]
[/vc_column_text][/vc_column][/vc_row]
8. Antibody dilutions
A guide for dilutions is usually provided with the antibody sheet, otherwise dilutions of 1 in 50 – 1 in 300 are good starting points for optimization; perhaps try a panel of primary antibody dilutions with a brand new antibody. Dilute the primary antibody in blocking solution (e.g. 3% BSA in PBS), then incubate for 1-2 hours at room temperature, or overnight at 4°C.
9. Washings
Wash off the primary antibody with wash solution such as PBS. A quick wash followed by three 5 minute washes should be sufficient. Frozen sections can be quite fragile, so drop solution slowly next to the section e.g. with a 1 ml pipette. Top tip: Alter the washing buffer to Tris-buffered Saline (TBS) rather than PBS.
10. Detection
Detection of the primary antibody is usually carried out with a secondary antibody directed against immunoglobulins of the host species of the primary antibody, conjugated to a fluorescent (e.g. FITC) or a chromogenic enzymatic tag (such as horse-radish peroxidase (HRP). Follow these tips for happy detecting!
- Background staining can vary between experiments, so it is a good idea to perform a negative control in which the primary antibody is omitted for each experiment.
- Dilute the secondary antibody in blocking solution, usually at around 1 in 800 – 1 in 1000 dilution (although this can be adjusted in future).
For fluorescence experiments:
- Check that the fluorophore you intend to use is detected by your microscope. It is important to avoid spectral overlap of the fluorochromes tagged to the secondary antibody, cross-reactivity between secondary antibodies and bleed-through of the signal whilst imaging.
- Keep sections in the dark once fluorescent antibodies have been added to prevent bleaching.
- After incubation, wash the sections several times with PBS before adding mounting medium to the sections and carefully lowering a coverslip over the top.
- Lower the coverslip gently to avoid air bubbles. Take particularly care with frozen sections, try not to squeeze or press the coverslip too hard as this may disrupt the tissue.
- It is often useful to stain the nuclei e.g. with DAPI, which can be included in a wash step or in the mounting medium.
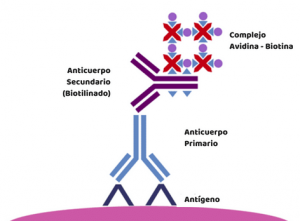
- It is possible to use primary antibodies directly conjugated to fluorescent tags. This saves time and reduces false signal but the level of signal will be lower. There are methods of amplifying low-level signals, including avidin-biotin-complex (ABC) and labelled streptavidin-biotin method (LSAB).
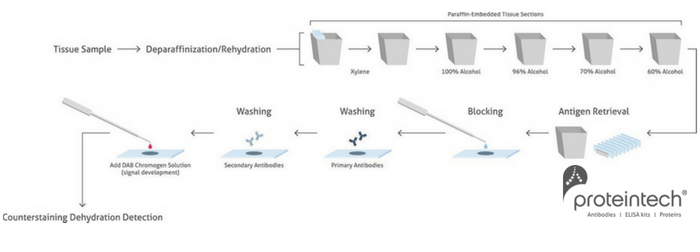
This is a summary of the main steps of the protocol:
¿Tienes dudas?
Si no te queda claro del todo cómo funciona esta tecnología, o quieres que te ayudemos a configurar tu ensayo, nuestro departamento técnico de especialistas, con amplia trayectoria en investigación (todos PhD), te pueden echar una mano: por mail (tecnic@labclinics.com), por tlf +34.934464700 o de forma presencial. Contáctanos y estaremos encantados de poder ayudarte!


Leave a reply