
The development of cancer is a multiphasic process in which healthy cells acquire characteristics that lead them to become tumor cells.
1) Introduction
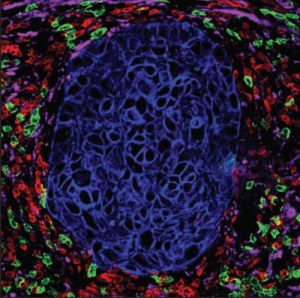 The tumor microenvironment (TME) consists of low [O2], low pH, and high concentrations of fatty acids. The high amounts of fatty acids are produced by cancer cells as a metabolic by-product. Myeloid-derived suppressor cells (MDSCs) thrive on these high fatty acid concentrations via fatty acid oxidation (FAO). Under these conditions, they support tumor growth by suppressing T cells that would normally attack the tumor.
The tumor microenvironment (TME) consists of low [O2], low pH, and high concentrations of fatty acids. The high amounts of fatty acids are produced by cancer cells as a metabolic by-product. Myeloid-derived suppressor cells (MDSCs) thrive on these high fatty acid concentrations via fatty acid oxidation (FAO). Under these conditions, they support tumor growth by suppressing T cells that would normally attack the tumor.
Although not their primary mechanism, certain chemotherapeutic agents inhibit FAO, sensitizing cells within the TME to death by starvation [1,2]. Though often difficult to simulate [3] i t i s u seful t o r eproduce the unique environment of the TME in vitro to give a better understanding of how cells behave in this environment. We demonstrate the ability to replicate the atmospheric conditions of the TME using a Seahorse XFe96 to create a tumor-relevant model. We also use a combination of compounds to inhibit transporters of the main mitochondrial fuel pathways (Figure 1). These compounds are used to examine the flexibility (capacity and dependency) of cancer cells’ and primary human MDSCs’ reliance on pyruvate oxidation versus glutamine metabolism versus FAO by performing a Mitochondrial Fuel Flex Test.
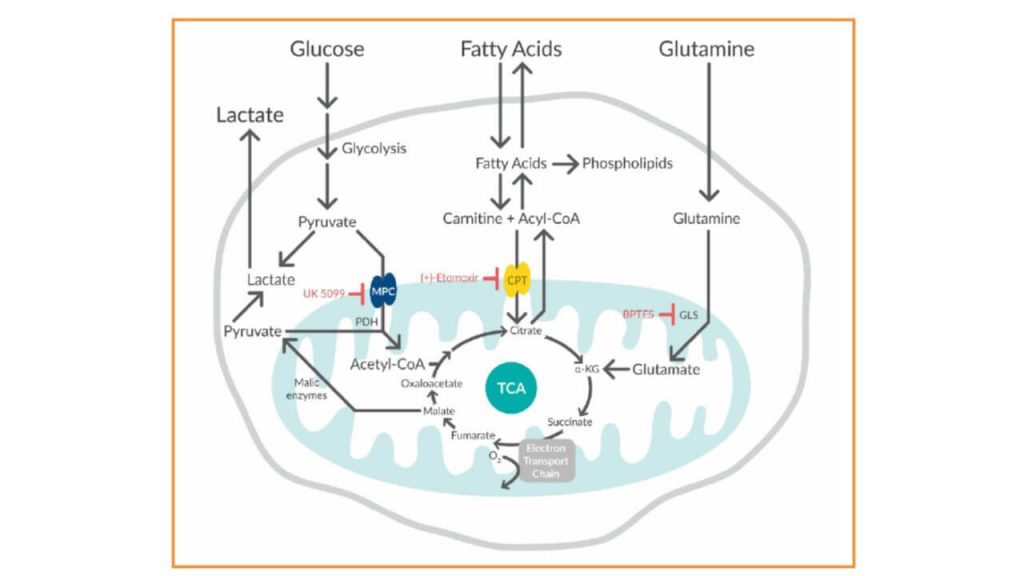
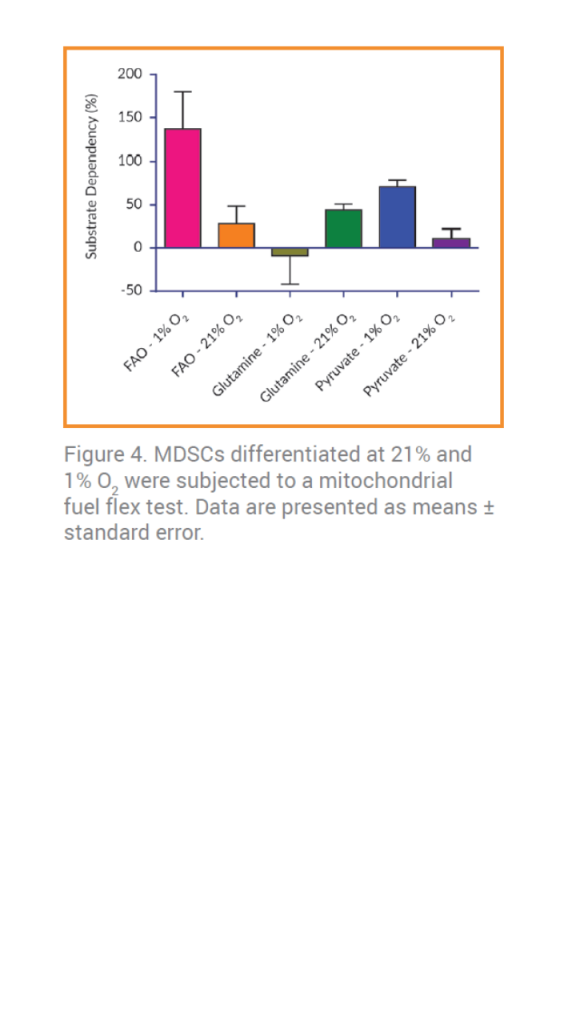
Figure 1. Three pathway inhibitors demonstrate the flexibility of cells to use glucose, glutamine, and fatty acids as mitochondrial fuel sources. MPC=mitochondrial pyruvate carrier; CPT=carnitine palmitoyltransferase; GLS=glutaminase.
2) Methods
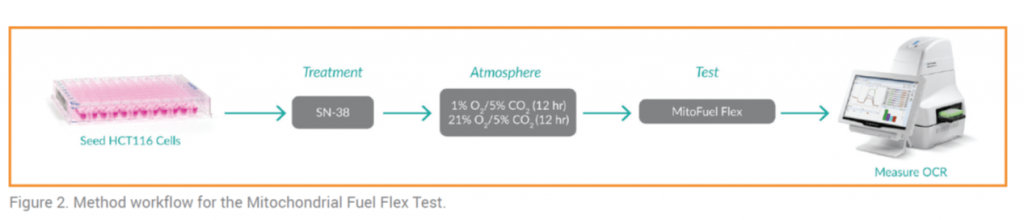
• HCT116 human colon carcinoma cell culture: XFe96 plates were treated with the chemotherapeutic agent SN-38 or vehicle and maintained at either ambient (21% O2/5% CO2) or low (1% O2/5% CO2) [O2] [O2] (Figure 2).
MDSC isolation: MDSCs were differentiated from monocytes isolated from whole blood. After 4 days, cells were placed at 1% O2/5% CO2 or maintained at 21% O2/5% CO2 for the remainder of the differentiation period (6 days total) before being analyzed.
• Mitochondrial Fuel Flex TestCapacity and dependency for FAO, glutamine, and pyruvate were determined through the injection of (+)-etomoxir (2 μM) to inhibit CPT1 (FAO), UK 5099 (4 μM) to inhibit MPC (mitochondrial pyruvate oxidation), BPTES (3 μM) to inhibit GLS (glutamine oxidation) Oxygen consumption rate (OCR) was measured using a Seahorse XFe96. OCR values at points of equilibrium were used to calculate a percentage of substrate utilization for pyruvate oxidation, glutamine metabolism, and FAO after exposure to various combinations of inhibitor sets.
3) Results
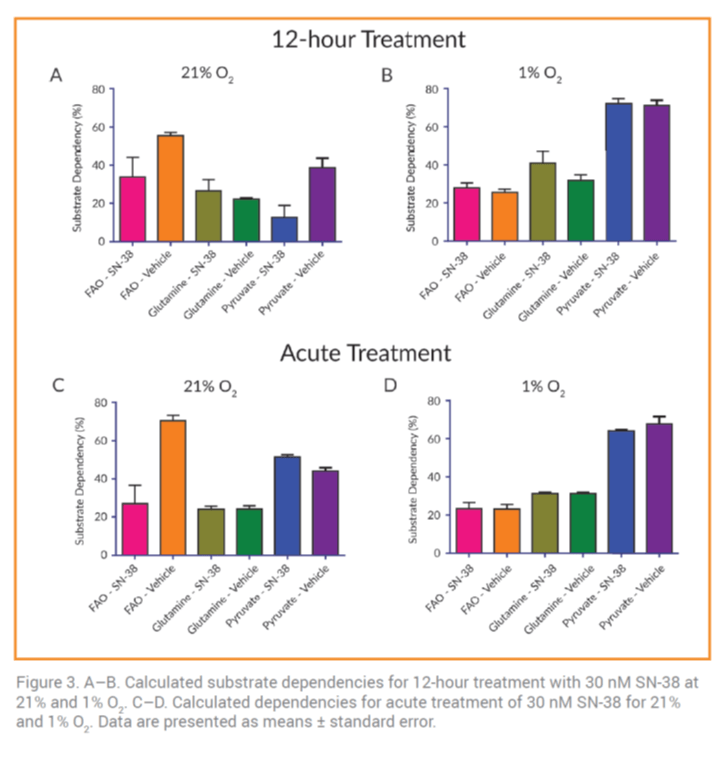 When HCT116 cells were treated with SN-38 with two types of treatment: acute or for 12 hours. The oxidation capacity of pyruvate, glutamine and FAO did not change significantly (data not shown). This suggests that under normal oxygen concentrations, HCT116 cells are more dependent on FAO. In contrast, at 1% [O2], acute SN-38 exposure increased the dependence of HCT116 cells on pyruvate (Figure 3B and 3D). This observation was expected since it increases glycolysis regulated by the availability of pyruvate.
When HCT116 cells were treated with SN-38 with two types of treatment: acute or for 12 hours. The oxidation capacity of pyruvate, glutamine and FAO did not change significantly (data not shown). This suggests that under normal oxygen concentrations, HCT116 cells are more dependent on FAO. In contrast, at 1% [O2], acute SN-38 exposure increased the dependence of HCT116 cells on pyruvate (Figure 3B and 3D). This observation was expected since it increases glycolysis regulated by the availability of pyruvate.
In contrast to this, MDSCs cultured at 21% [O2]O2 appear to have increased dependency on glutamine (Figure 4). At 1% O2, however, a shift in substrate dependency in favor of FAO was observed. This switch in substrate dependencies at 21% O2 compared to 1% O2 suggests that lower [O2] alters the metabolic phenotype of MDSCs.
4) Conclusions
Culture conditions, particularly oxygen concentration, can have a significant impact on metabolic substrate utilization. The ability to assess mitochondrial fuel flexibility in a controlled environment using a combination of inhibitors that pinpoint specific metabolic pathways presents the opportunity to observe cells under conditions that are a better representation of a native environment.
 With a critical understanding of how substrate utilization and metabolic activity are reprogrammed, new strategies can be developed to modulate the enzymes and transporters driving mitochondrial oxidation pathways. Knowing the factors that affect the dysregulation of glucose, lipid, and/or amino acid homeostasis during diseases such as diabetes, obesity, fatty liver diseases, mitochondrial disorders, cardiac failure, neurodegeneration, and cancer will improve the discovery of targeted therapeutics.
With a critical understanding of how substrate utilization and metabolic activity are reprogrammed, new strategies can be developed to modulate the enzymes and transporters driving mitochondrial oxidation pathways. Knowing the factors that affect the dysregulation of glucose, lipid, and/or amino acid homeostasis during diseases such as diabetes, obesity, fatty liver diseases, mitochondrial disorders, cardiac failure, neurodegeneration, and cancer will improve the discovery of targeted therapeutics.
5) References
1. Schumacher, J.D. and Guo, G.L. Mechanistic review of drug-induced steatohepatitis. Toxicol. Appl. Pharmacol. 289(1), 40–47 (2015).
2. Begriche, K., Massart, J., Robin, M.A., et al. Drug-induced toxicity on mitochondria and lipid metabolism: Mechanistic diversity and deleterious consequences for the liver. J. Hepatol. 54(4), 773–794 (2011).
3. Vander Heiden, M.G. Exploiting tumor metabolism: Challenges for clinical translation. J. Clin. Invest. 123(9), 3648–3651 (2013).






Leave a reply